Week 2 - Genomics and Data Analysis
What is genome annotation?
After reading a nucleotide sequence, all we have is a raw string of nucleotides (A, T, C, and G). To generate insight into the function, location, and regulation of genes, biologists add notes to specific segments of the genome to attach important biological information to various segments of the genome. In addition to locating genes, these notes serve to pinpoint regulatory elements that control gene expression, such as promoters and enhancers, and non-coding regions. This process is called genome annotation.
Why is genome annotation important?
Genome annotation is a process that allows us to understand what is inside a genome and how it works. This leads to important applications in numerous fields, including:
Understanding Diseases: Helps in identifying genes involved in diseases, leading to better diagnostics and treatments.
Agriculture: Improves crop and livestock by identifying beneficial genes.
Evolutionary Biology: Helps trace the evolution of species by comparing genomes.
Biotechnology: Facilitates the development of new products like biofuels and pharmaceuticals.
What are the steps of genome annotation?
1. Repeat masking
Mask repetitive elements in the genome (e.g., TTTTTT) to prevent interference with subsequent steps.
2. Evidence alignment
Match the genome with sequence data (proteins, RNA, etc.) from other experiments to help determine where genes and other elements are located.
3. Feature Prediction
Use computational tools to identify the regions of DNA that are likely to encode proteins, as well as regulatory elements and non-coding regions.
4. Functional annotation
Predict the potential function of the identified genes and non-coding regions.
Repeat masking
Believe it or not, repetitive sequences like CCCCCCCCCCC or duplicated genetic information represent a major portion of many genomes, including the human genome. These sequences must be masked (replaced with a character different than A, C, G, or T) to prevent interference with the identification of genetic features of interest, such as protein-coding genes.
In order to mask repeats, researchers use a repeat library for the particular genome being annotated. Tools like RepeatMasker detect repeats in the genome using the repeat library and mask them either with a different character (like R) or with a lower case version of the original nucleotide sequence (using a, c, g, or t).
Evidence alignment
Sequence data from other experiments can serve as clues to where genes are. Specifically, biologists align all known RNAs and proteins of the organism with it’s genome. Since RNA and protein sequences can be translated from DNA and vice versa, biologists are able to determine where sites of corresponding DNA might be.
In addition to RNA and protein, expressed sequence tags (EST) are aligned with the genome. Expressed sequence tags are short pieces of DNA that come from genes that are actively being used by cells. By matching these tags with the genome sequence, scientists can figure out where genes are located and how they are structured.
As you may recall, BLAST (Basic Local Alignment Search Tool) is a widely used tool for comparing a query sequence against a database of known sequences to find similar sequences and can be used for evidence alignment.
Feature prediction
Feature prediction is step in which genomic segments are identified as regions of DNA that are likely to encode proteins, as well as regulatory elements and non-coding regions. Like the previous steps, a variety of computational tools are used.
Protein-coding gene prediction
There are three main approaches for predicting which parts of the genome code for proteins:
Intrinsic (ab initio): Uses information from the genomic sequence itself. It requires building and training specific models for each genome.
Extrinsic: Uses existing data like protein sequences and transcripts from other species. It relies on databases and alignment techniques.
Combiners: Combines both intrinsic and extrinsic methods, often giving the best results. An example of a combiner is MAKER.
Regulatory element prediction
Tools like MEME Suite identify regulatory elements such as promoters, enhancers, and transcription factor binding sites using statistical or machine learning algorithms to recognize sequence patterns associated with different types of regulatory elements.
Non-coding RNA prediction
Computational tools identify non-coding RNA genes such as tRNAs, rRNAs, and miRNAs. tRNAscan-SE, for example, is used to predict tRNA genes, while Infernal can be used to identify other non-coding RNAs based on covariance models.
Functional annotation
Once the genes are identified, their potential functions are predicted. Researchers use a standard vocabulary to systematically classify the functions of genomic segments. This system is called gene ontology (GO) and it has three functional categories: Molecular function, biological process, and cellular component.
Graphs can represent gene ontology, with each GO term as node in a roughly hierarchical structure. The relationship between the terms are described the arrows, with “parent” terms being broader classifications than their “child” terms.
The relationships between genes represented by GO changes as discoveries in genomics lead researchers to new understandings of the functions of genes. How are such discoveries made? There are multiple approaches to identify the functions of genes:
Homology-based methods rely on similarities with another genome to infer similar functions. Researchers use BLAST searches to find these similarities.
Probabilistic methods analyze amino acid distributions to predict function, such as determining if a protein is soluble or membrane-bound.
Machine learning algorithms are trained to associate genetic data patterns with certain GO terms

A gene ontology graph represents the relationships between functional categories. Image credit: GO Consortium
Functional annotation of noncoding sequences, such as pseudogenes, segmental duplications, and RNA genes, involves specific methods tailored to each type.
Pseudogenes
Background: Pseudogenes are genetic sequences that resemble functional genes but have lost their ability to produce functional proteins or RNA molecules. They are often considered non-functional relics of once-functional genes due to mutations or genomic rearrangements that have rendered them inactive.
PseudoPipe: Identifies and characterizes pseudogenes by aligning protein sequences to the genome and detecting features like lack of introns or presence of frameshifts.
Ensembl Pseudogene Module: Detects processed pseudogenes by looking for single-exon copies of multi-exon genes and other characteristics like intronic repeats.
Segmental duplications
Background: Segmental duplications are substantial regions of the genome that have been duplicated from one location to another. These duplicated segments typically range in size from 1,000 to 200,000 base pairs
BLAST: Aligns genomic sequences against themselves to find large similar regions indicative of segmental duplications.
RNA genes
Background: RNA genes are segments of DNA that are transcribed into RNA molecules but are not translated into proteins. Instead, the RNA molecules perform various essential functions in the cell such as aiding in protein synthesis, RNA processing, and regulating gene expression.
Infernal and Rfam: Predict small RNAs (sRNAs) using covariance models and RNA family databases.
Ensembl Annotation System: Incorporates methods to annotate various types of RNA genes, though specific tools are not detailed.
How computational biology uses genome annotation
Comparative genomics: Annotated genomes allow for comparisons between species, helping researchers study the evolution of genomes.
Pathway and network analysis: Functional annotation of genes enables the reconstruction of metabolic pathways and regulatory networks.
Disease gene identification: Annotation helps identify potential disease-causing genes by providing context for interpreting genomic variations.
Metagenomics: Annotation helps identify and characterize genes from multiple organisms in environmental samples.
Transcriptomics and proteomics: Genome annotation provides the reference framework for analyzing gene expression data and protein interactions.
Epigenetics research: Annotation of regulatory elements and non-coding regions aids in interpreting epigenetic modifications and their impacts.
Bacterial Gene Annotation Activity
In this activity, you will learn to use some web-based genome annotation tools to:
Perform sequence alignment to identify the protein encoded within a given sequence
Locate the coding region of the protein
Locate the promoters within the sequence
Determine the function of the sequence within the context of the genome
Let’s get started! Here is the DNA sequence we will be working with:
GTAGTGTGTTCCACTGCTGATTTCTTTTTGAGCATTATAGGCGGTATGTTGGAAGAAAAGAACAAAAGTTTGTGAACGATAGCATATAGGTATTAAGAATACGATGAACTCGTTGGTAATATTGACGTACTCAAGTGCGTACGCTATAATTAGCGTAACAGAAAACAAAGGAGGGTTTATATGATTATCACTAGCCCTACAGAAGCGAGAAAAGATTTTTATCAATTACTAAAAAATGTTAATAATAATCACGAACCAATTTATATTAGTGGCAATAATGCCGAAAATAATGCTGTGATTATAGGTTTAGAAGATTGGAAAAGTATACAAGAGACAATATATCTTGAATCTACTGGTACAATGGACAAAGTAAGAGAAAGAGAAAAAGATAATAGTGGTACAACAAATATAGATGATATTGATTGGGATAATCTTTAATGAGCAATTACACGGTTAAGATTAAAAATTCAGCGAAATCAGATTTAAAGAAAATAAAACATTCTTATTTAAAGAAGTCATTTTTAGAAATTGTTGAGACTTTAAAAAATGATCCGTATAAAATAACACAATCTTTTGAAAAATTAGAGCCTAAATATTTAGAGCGATATTCAAGAAGAA
Copy the sequence (ctrl + c). Then, follow the following steps to annotate the sequence. The solution to each step can be found at the bottom of this page.
1. Alignment using BLAST
For simplicity sake, the provided DNA sequence does not include any repeats. Thus, we will begin with the second step of genome annotation, evidence alignment. In this step we will be using BLAST to see if a database of sequences data has any matches with our sequence. This will provide evidence for a protein-encoding gene and indicate a the protein encoded. Navigate to https://blast.ncbi.nlm.nih.gov/Blast.cgi and select “blastx”. Paste in the sequence and click “BLAST”. Be patient while waiting! It is searching an entire database, and normally takes around 30 seconds.
Based on the results, what protein is likely encoded within the sequence given? From what species of bacteria is it derived?
2. Feature prediction
In this step, we will be predicting the location of the protein-encoding gene, as well as promoters, in our sequence. First, we will annotate the gene.
To figure out where the gene is, we need to choose the open reading frame most likely to contain the gene. An open reading frame is the sequence of codons (three base pairs that together code for a single amino acid) between the start codon (AUG) and a stop codon (TAA, TAG, or TGA). There are three possible reading frames, since a codon has three base pairs (you can shift your reading of codons two times before arriving at the original reading frame). Let’s use an online tool to quickly find the longest reading frame, the likely location of the protein-encoding gene.
Navigate to https://web.expasy.org/translate/. Paste the provided DNA sequence into the text box and choose the default “verbose” format from the pull down menu below the box. Click on "Translate sequence". You will notice that the program translates the DNA sequence into 6 different reading frames. The first three results are in the forward direction and bottom three are in the reverse direction (as if you read the base pairs starting from the end).
The tool should highlight the open reading frames in red. Identify the longest open reading frame and which reading frame it comes from (1-6). This is the location of the protein-encoding gene we identified in step 1.
Now, let’s find the promoters in our sequence. In many bacteria, including the species from which the provided DNA sequence was derived, the promoters that help cellular machinery transcribe DNA into RNA has two short sequences before the gene. These sequences are called “consensus” sequences.
Navigate to http://www.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb. Paste the provided DNA sequence into the text box and click “Process”.
The computer output from the tool should predict two potential promoters. Here’s how to interpret to computer output:
One of these predicted promoters is after the open reading frame we found as the likely location of the gene. Now consider, the other promoter. It should predict a “-10 box” and a “-35 box”. This means that there are two parts of the promoter, one 10 nucleotides before the protein, and one 35 nucleotides before the protein. These are called the promoter boxes. Does the location of this promoter make sense in relation to the location of our predicted gene?
3. Functional annotation
The third and final step of this activity is to determine the function of the protein we identified. When a new protein-encoding gene is discovered, the variety of methods described in this lesson’s explanation of functional annotation are used to predict the new protein’s function. However, these methods are beyond the scope of this activity. Additionally, we already identified the protein in step 1. Therefore, let’s use a gene ontology lookup service at https://www.ebi.ac.uk/ols4/ to classify the gene’s function. Paste the name of the gene we found in step 1 and click “search”. Click on the first two results and examine the functions described. What can we now say about the protein’s function?
Solutions
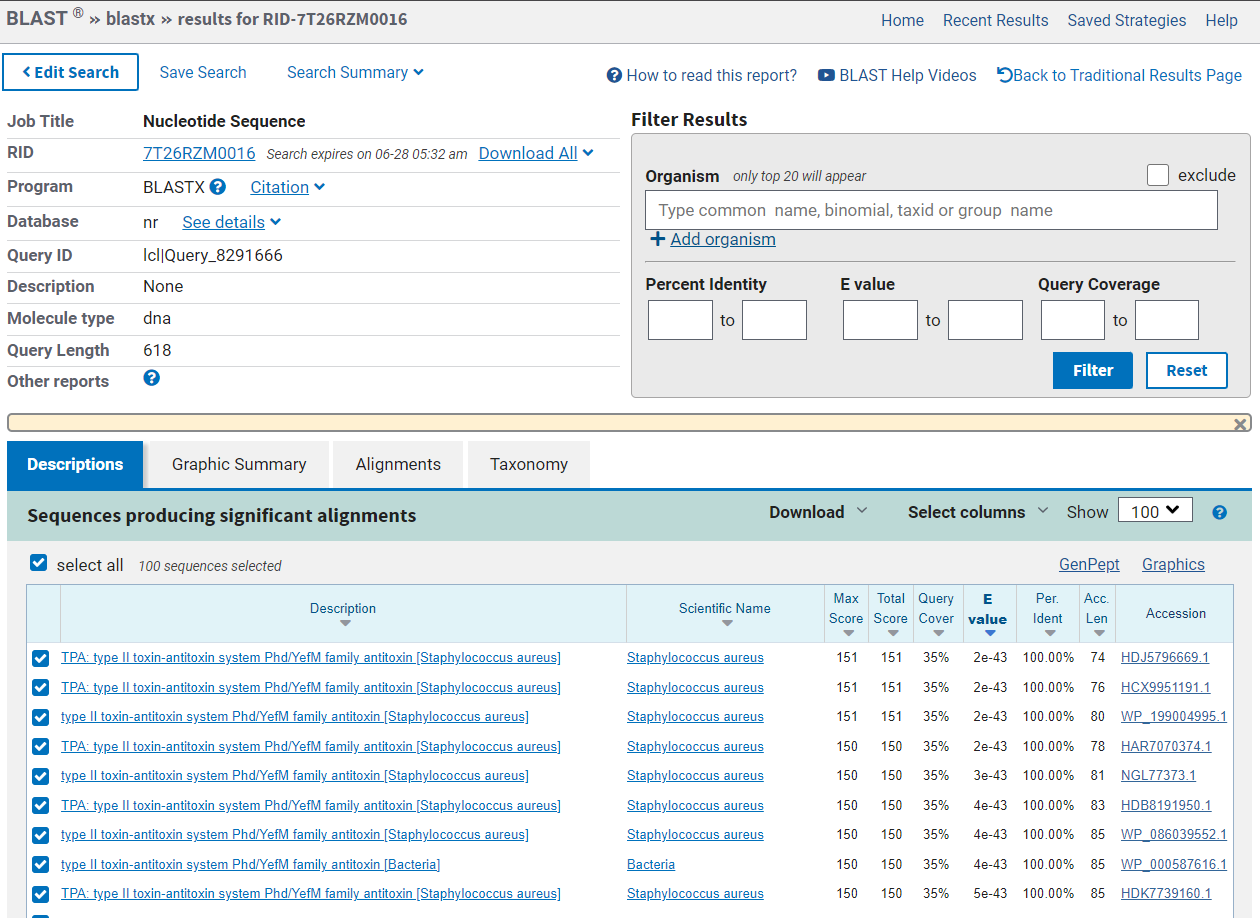
After navigating to BLAST and entering your sequence, you should have the following view:
Based on the search results, you should have identified the protein as type II Phd/YefM family antitoxin of the bacteria Staphylococcus aureus.
The output of the expasy tool should look like this:
As you can see, the longest open reading frame is in frame 1. This is the likely location of the protein-encoding gene.
The output of the promoter-finding tool should look like this:
The computer output indicates that there are two plausible promoters, one at 157 nucleotides and one at 467 nucleotides. The one at the 467 nucleotide position is after the open reading frame we found to be the likely position of the gene; therefore, we can rule it out as a potential promoter. The promoter at 157 nucleotides is in the correct position (before the predicted gene location).
Here is the result of the GO search:
The first two search results tell us the following about the protein’s function:
The protein has the molecular function of binding to toxins to to prevent it from interacting with other partners
As a cellular component, it forms a toxin-antitoxin complex to regulate it’s partner toxin’s activity
This activity was adapted from an activity used in the BSCI 222 course at the University of Maryland. The source activity can be accessed via this link.